Molecular dynamics simulation sheds new light on methane hydrate formation

In a paper published this week in PNAS, researchers at the University of Amsterdam's Van 't Hoff Institute for Molecular Sciences and the Amsterdam Center for Multiscale Modeling provide atomistic insight in the formation of methane hydrates. On the basis of molecular dynamics simulations they explain how selection between competing methane hydrate polymorphs occurs, and how this might be generalized to other hydrates and molecular crystal formation.
Methane hydrate are ice-like solid substances that are abundantly present, among others at the ocean floors. It is estimated that the amount of energy stored in methane hydrates is twice the amount of energy stored in conventional resources of fossil fuels. At the same time, the formation of hydrates is of concern to the petroleum industry as they can clog oil pipelines, causing flow problems. Methane hydrates are also present in the permafrost in arctic regions. The thawing of the permafrost as a result of increasing global temperatures can lead to the release of large amounts of methane, which is a powerful greenhouse gas.
Encaged methane molecules
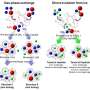
In a methane hydrate, at the molecular level methane is encaged inside a hydrogen-bonded water network. While methane gas is hydrophobic under ambient conditions, at low temperatures and high pressures a mixture of water and methane gas can spontaneously nucleate into hydrates.
Over the years, the interest in comprehending the formation mechanism of hydrates has increased tremendously. In particular their formation under natural conditions is poorly understood. Understanding the process of homogeneous nucleation, and how this leads to different methane hydrate polymorphs, can lead to improved control of crystallization, as well as insight into polymorph selection in general.
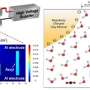
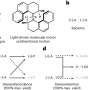
, 280 K (red), and 285 K (green). The arrows indicate schematically pathways traversing from liquid to solid (dashed arrows: to amorphous phase; solid arrows: to crystalline phase). While at low temperatures (e.g., 275 K), the free energy barrier to nucleate the amorphous solid is lowest, the trend is reversed at higher temperature (e.g., 285 K), where sampled pathways mostly end up in the crystalline phase. At 280 K both mechanisms are accessible. Credit: HIMS/PNAS")
Novel simulation approach
Since experimental research on the formation of the different methane hydrate polymorphs suffers from limited resolution, the Amsterdam researchers led by professor Peter Bolhuis used molecular dynamics simulations to provide such insight.
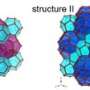
Applying a direct molecular dynamics simulation is not very effective, because at moderate undercooling nucleation is a very rare event, due to the presence a very high energy barrier. Such a simulation would require computation times beyond the age of the Universe. However, because the nucleation event in itself, while rare, occurs very fast (on a microsecond time scale), the researchers could create a large collection of molecular dynamics trajectories that exhibit these fast events. Subsequent detailed analysis of these trajectories showed how selection between competing amorphous and crystalline polymorph formation mechanisms takes place. Their PNAS paper not only sheds light on the formation of methane hydrates, but also on other clathrate compounds and molecular crystal formation in general.
More information: undefined Arjun et al. Unbiased atomistic insight in the competing nucleation mechanisms of methane hydrates, Proceedings of the National Academy of Sciences (2019). DOI: 10.1073/pnas.1906502116
Journal information: Proceedings of the National Academy of Sciences
Provided by University of Amsterdam