How to measure a molecule's energy using a quantum computer
 – the largest molecule simulated on a quantum computer to date. The results demonstrate a path of exploration for near-term quantum systems to enhance our understanding of complex chemical reactions that could lead to practical applications. Credit: Kandala et al.; Nature")
Simulating molecules on quantum computers just got much easier with IBM's superconducting quantum hardware. In a recent research article published in Nature, Hardware-efficient Variational Quantum Eigensolver for Small Molecules and Quantum Magnets, we implement a new quantum algorithm capable of efficiently computing the lowest energy state of small molecules. By mapping the electronic structure of molecular orbitals onto a subset of our purpose-built seven qubit quantum processor, we studied molecules previously unexplored with quantum computers, including lithium hydride (LiH) and beryllium hydride (BeH2). The particular encoding from orbitals to qubits studied in this work can be used to simplify simulations of even larger molecule and we expect the opportunity to explore such larger simulations in the future, when the quantum computational power (or "quantum volume") of IBM Q systems has increased.
While BeH2 is the largest molecule ever simulated by a quantum computer to date, the considered model of the molecule itself is still simple enough for classical computers to simulate exactly. This made it a test case to push the limits of what our seven qubit processor could achieve, further our understanding of the requirements to enhance the accuracy of our quantum simulations, and lay the foundational elements necessary for exploring such molecular energy studies.
The best simulations of molecules today are run on classical computers that use complex approximate methods to estimate the lowest energy of a molecular Hamiltonian. A "Hamiltonian" is a quantum mechanical energy operator that describes the interactions between all the electron orbitals and nuclei of the constituent atoms. The "lowest energy" state of the molecular Hamiltonian dictates the structure of the molecule and how it will interact with other molecules. Such information is critical for chemists to design new molecules, reactions, and chemical processes for industrial applications.
Qubit:Orbital
Although our seven qubit quantum processor is not fully error-corrected and fault-tolerant, the coherence times of the individual qubits last about 50 µs. It is thus really important to use a very efficient quantum algorithm to make the most out of our precious quantum coherence and try to understand molecular structures. The algorithm has to be efficient in terms of number of qubits used and number of quantum operations performed.
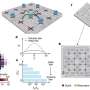
, exact energy surfaces (dotted lines) and density plots (shading; see colour scales) of outcomes from numerical simulations, for several interatomic distances for H2 (a), LiH (b) and BeH2 (c). The experimental and numerical results presented are for circuits of depth d = 1. The error bars on the experimental data are smaller than the size of the markers. The density plots are obtained from 100 numerical outcomes at each interatomic distance. The top insets in each panel highlight the qubits used for the experiment and the cross-resonance gates (arrows, labelled CRc–t; where ‘c’ denotes the control qubit and ‘t’ the target qubit) that constitute UENT. The bottom insets are representations of the molecular geometry (not to scale). For all the three molecules, the deviation of the experimental results from the exact curves is well explained by the stochastic simulations. Credit: Kandala et al.; Nature")
Our scheme contrasts from previously studied quantum simulation algorithms, which focus on adapting classical molecular simulation schemes to quantum hardware – and in so doing not effectively taking into account the limited overheads of current realistic quantum devices.
So instead of forcing classical computing methods onto quantum hardware, we have reversed the approach and asked: how can we extract the maximal quantum computational power out of our seven qubit processor?
Our answer to this combines a number of hardware-efficient techniques to attack the problem:
- First, a molecule's fermionic Hamiltonian is transformed into a qubit Hamiltonian, with a new efficient mapping that reduces the number of qubits required in the simulation.
- A hardware-efficient quantum circuit that utilizes the naturally available gate operations in the quantum processor is used to prepare trial ground states of the Hamiltonian.
- The quantum processor is driven to the trial ground state, and measurements are performed that allow us to evaluate the energy of the prepared trial state.
- The measured energy values are fed to a classical optimization routine that generates the next quantum circuit to drive the quantum processor to, in order to further reduce the energy.
- Iterations are performed until the lowest energy is obtained to the desired accuracy.
With future quantum processors, that will have more quantum volume, we will be able to explore the power of this approach to quantum simulation for increasingly complex molecules that are beyond classical computing capabilities. The ability to simulate chemical reactions accurately, is conductive to the efforts of discovering new drugs, fertilizers, even new sustainable energy sources.
The experiments we detail in our paper were not run on our currently publically available five qubit and 16 qubit processors on the cloud. But developers and users of the IBM Q experience can now access quantum chemistry Jupyter notebooks on the QISKit github repo. On the five qubit system, users can explore ground state energy simulation for the small molecules hydrogen and LiH. Notebooks for larger molecules are available for those with beta access to the upgraded 16-qubit processor.
More information: Abhinav Kandala et al. Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets, Nature (2017). DOI: 10.1038/nature23879
Journal information: Nature
Provided by IBM